Ataxia telangiectasia (A-T) [OMIM #208900] is an autosomal recessive multisystem disorder that is typically characterized by progressive cerebellar ataxia, oculocutaneous telangiectasia, variable immunodeficiency, increased alpha-fetoprotein levels, and hypersensitivity to ionizing radiations. A-T occurs at a frequency ranging from one in 40 000 to one in 100 000 births worldwide and ATM [OMIM *607585], an important player in DNA damage response/repair, is the only gene known to be associated with this disease.1
There is a wide spectrum of phenotypic manifestations in patients with A-T. Recently, there is growing evidence that ATM mutations, can manifest generalized or focal dystonia with or without of the classical signs of A-T.2,3 Here, we report a patient with classical A-T phenotype, hyper immunoglobulin (IgM) immune phenotype, and generalized dystonia, and literature review of reported cases with dystonia.
Case report
The 10-year-old girl was born to non-consanguineous, healthy, Iranian parents after an uneventful gestation and delivery. Her older sister was healthy, and there was no family history of neurological disease or malignancy. The first symptom that was noticed by parents was repeated diarrhea, which started when she was six months old until she turned two years old. At that time, inflammatory bowel disease was diagnosed by physicians based on diarrhea, failure to thrive and underweight, and the results of laboratory tests. She walked at about 16 months old, but after eight months (aged two years old), she manifested mild gait ataxia (abnormal swaying of the head and trunk). Her walk has improved since her parents started occupational therapy and her speech, fine motor and social skills were normal, but diarrhea continued. By the age of five years, cerebellar ataxia had progressed, and her speech had become dysarthric. At seven years old, when she was referred to our hospital, she had lost the ability of independent ambulation, and ocular telangiectasia was evident. Diagnosis of A-T was made based on clinical and laboratory findings and she was started on intravenous immunoglobulin therapy (IVIG).
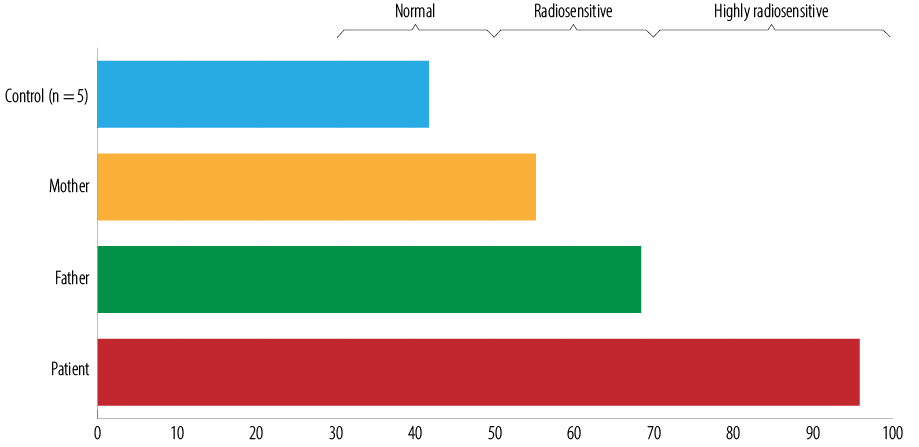
Figure 1: G2-chromosomal radiosensitivity assay results in a patient with ataxia telangiectasia and her parents. Individual radiosensitivity (IRS) estimated as IRS = [1-(G2caf-G2)/G2caf] × 100%.
Laboratory finding showed serum IgG 5 mg/dL (normal: 800–1600), IgM 208 mg/dL (normal: 40–230), IgA 1 mg/dL (normal: 70–400), IgE 1 mg/dL (normal: up to 52); serum alpha-fetoprotein (AFP) levels 104.7 IU/mL (normal: < 5.5); and increased induced radiosensitivity as assessed by the G2 chromosomal radiosensitivity assay [Figure 1].
When the patient was 10-years-old, she was no longer able to walk and received granulocyte-colony stimulating factor (G-CSF, two times) and antibiotic prophylaxis due to neutropenia and lymphadenopathy granulomatous. During this year, she experienced brief episodes of involuntary head extension and rotation, and her cervical dystonia worsened and extended to the limbs and trunk, mostly on the right side, causing an abnormal posture. Brain magnetic resonance imaging showed mild cerebral atrophy, mainly in the lower cerebellar hemisphere. The cerebellar vermis was intact, whereas the fourth ventricle showed some dilation. Therapeutic trials using trihexyphenidyl HCl and subsequently baclofen were initiated but were both ineffective. However, trihexyphenidyl with clonazepam relatively reduced the symptoms.
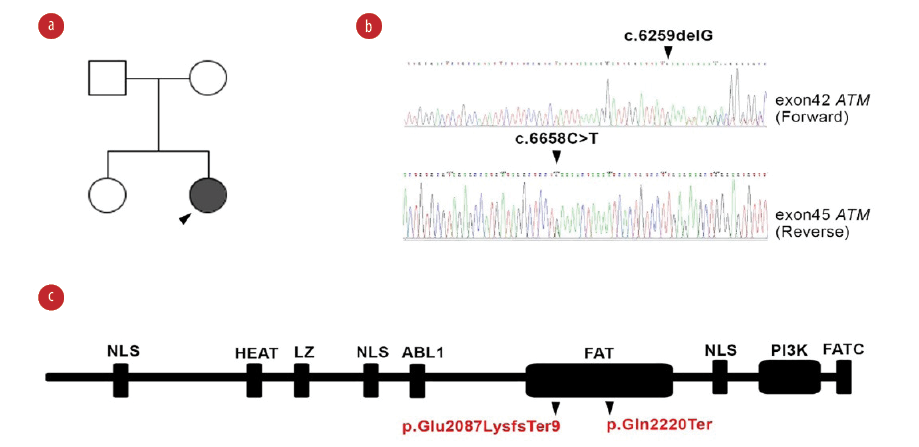
Figure 2: Genetic study of ATM in ataxia telangiectasia patient. (a) Pedigree of the patient. (b) Confirmatory Sanger sequencing of proband for identified variants of the ATM gene. (c) Representation of mutation on ATM protein domains showing defect on FAT domain at amino acids region 1960 to 2566.
Whole exome sequencing identified a compound heterozygous mutation in the ATM gene (c.6259delG and c.6658C>T) and was confirmed by Sanger sequencing in the patient and her parents. Both variants were located in the FAT domain (the name derived from FRAP, ATM, and TRRAP) of the ATM protein (p.Glu2087LysfsTer and p.Gln2220Ter; [Figure 2]). Bioinformatical tools support the pathological involvement of these variants (frequency: 0 in all databases, combined annotation-dependent depletion (CADD) scores: 35.00 and 45.00, respectively, and the mutation significance cutoff of ATM: 0.001).
Discussion
Biallelic mutations in ATM are associated with classical and milder forms (variant A-T) of A-T. The variant A-T does not present the cardinal features of the disease, or they become apparent only later in life with incomplete, atypical, and highly variable manifestations.2,4 The neurological aspects of A-T are unfortunately still poorly understood, and is the main aspect of the disease and is not recapitulated in mice with complete deficiency of the murine ATM ortholog.1 Impairment of the extrapyramidal movement system with signs including dystonia, myoclonus, chorea, parkinsonism, and both postural, rest, and kinetic tremor is also common among patients with A-T.2,3 Recently, reports of different types of dystonia related to ATM gene mutations have attracted great attention [Table 1, 2, and 3].
Table 1: Dystonia frequency in classic and variant ataxia telangiectasia (A-T).
|
19925 |
70 (2–42) |
Classic (62 cases) and variant (8) |
78% (55/70) |
Limbs, face, trunk, oromandibular |
|
20092 |
13 (NA) |
Variant |
72% (8/11) |
- |
|
20116 |
57 (2–19) |
Classic |
15.8% (9/57) |
- |
|
20147 |
22 (2.7–19.7) |
Classic |
50% (10/20) |
- |
Table 2: Dystonia reports in ataxia telangiectasia patients without ATM mutation analysis.
|
19689 |
1 |
C |
H |
Early (19) |
NA |
14 m |
NA |
NA |
IgA-L |
NA |
|
198010 |
1 |
C |
N, T, UL, LL |
Early
(9) |
N |
< 9 y |
NA |
High |
IgA-L |
NA |
|
198411 |
1 |
V |
G |
Early (16) |
N |
16 m |
NA |
normal |
IgE-L |
NA |
|
198712 |
2 |
V |
Dystonia |
Early
(< 16) |
NA |
NA |
NA |
Slightly high |
Normal |
Yes (CSA, ICBA) |
|
V |
UL |
Late (28) |
NA |
12 m |
NA |
Slightly high |
IgM-H |
Yes (CSA, G2A) |
|
198913 |
2 |
V |
L |
Early (7) |
Left hand |
Upon walking |
Atrophy (20 y) |
Slightly high |
Normal |
- |
|
V |
Dystonic gait |
Early (7) |
N |
4 y |
NA |
High |
IgA-L |
Yes (CSA, ICBA) |
|
199314 |
1 |
C |
LL |
Early
(~ 15) |
Left arm |
6 y |
Atrophy (17 y) |
High |
IgA-L |
NA |
|
199315 |
3 |
C |
UL, N |
Early
(~ 2) |
ULs |
~ 2 y |
Normal (1, 20 m) |
High |
IgA-L |
Yes (ICBA) |
|
199416 |
1 |
C |
CC, T, UL, F |
Early
(4) |
N |
2 y |
Atrophy (6.5 y) |
High |
IgA-L |
Normal (ICBA) |
Slightly high: 11-30, high: < 30 according to Ball and Waldmann studies.18,19
MN assay: S-G2 micronucleus assay, ICBA: induced chromosome breakage assay; CSA: colony survival assay; NA: not available; Ig: immunoglobulin
N: neck; T: trunk; UL: upper limb; LL: lower limb; CC: craniocervical; F: face; O: oromandibular; G: generalized; C: classic; V: variant; L: limbs; H: hand; RUL: right upper limb.
Dystonia is a movement disorder characterized by involuntary sustained or intermittent muscle contractions producing abnormal postures, repetitive movements, or both. Dystonic movements are usually manifested by twisting and may show a tremulous pattern. Before designating A-T as a distinct disease, dystonia was reported by Syllaba and Henner in three adolescent Czech siblings in association with ocular telangiectasia.34 After this report, the non-primary (secondary) dystonia35 was described in other articles repeatedly before
[Table 2] and after [Table 3] identification of the ATM gene in patients with A-T. The dystonia frequency in patients with A-T vary in different case series [Table 1], and since myoclonic dystonia can easily be mistaken for chorea, the exact frequency is not known, but at least in variant A-T it accounts for 86% of cases8 with prominent cervical, cranial, and brachial involvement [Table 1, 2, and 3]. Although dystonia has been manifested in both classic A-T and variant A-T at different ages with different body involvements, it has received great attention in the variant form, as dystonia is more frequent in this form of the disease and may in fact be the only symptom in variant A-T.32 Currently, the treatment of dystonia in A-T patients remains symptomatic [Table 4]. Due to the marked heterogeneity of A-T, we have limited knowledge about the basis of individual responses to a given therapy.
Dystonia, like other extrapyramidal features, is usually more prominent in late manifestations of classical A-T.6 However, it may also be the initial or even the most prominent manifestation of A-T,5,10 potentially masking ataxia symptoms.10 This symptom mostly appears as an early-onset form of dystonia, and in variant forms without frank cerebellar involvement.10,12,21,22,26–28,32 Therefore, it may mimic other forms of early-onset primary torsion dystonias22 or present mildly without any notice.32 In contrast to what we observed in our patient, dystonia is often associated with myoclonic jerks8,10,22,27,30 and may be induced/worsened by specific motor or cognitive tasks and, similar to ataxia, it is progressive with age as expected in a neurodegenerative disorder.6
Table 3: Dystonia reports in ataxia telangiectasia cases with confirmed ATM mutation.
|
200820 |
V |
1 |
N, LL |
Early (14) |
N |
14 |
Atrophy (42) |
Slightly high |
Normal |
NA |
|
200921 |
V |
1 |
CC, O |
Early (15) |
N |
No ataxia |
Mild atrophy (18) |
High |
NA |
Normal |
|
2009–201422–24 |
V |
12 |
N, L, C* |
Early (~ 12) |
Cervical |
Late-onset mild ataxia |
Mild atrophy (NA) |
High |
NA |
NA |
|
201325 |
V |
3 |
CR |
Early (~ 13) |
Cervical |
No ataxia |
NA |
High |
NA |
NA |
|
201326 |
V |
1 |
F, N |
Early (~ 7) |
NA |
No ataxia |
Normal (16) |
High |
IgA-Low |
Yes (MN assay) |
|
201327 |
V |
1 |
L, N, T |
Early (2) |
L |
No ataxia |
Normal (48) |
High |
NA |
Yes (ICBA) |
|
201328 |
V |
3 |
N, L, T |
Early (NA) |
Cervicobrachial |
No ataxia |
Normal (NA) |
High |
NA |
Yes (CSA) |
|
201329 |
V |
1 |
Dystonia |
Early (NA) |
NA |
No ataxia |
Atrophy (41) |
High |
IgA-Low |
Yes (CSA) |
|
201430 |
V |
1 |
LL, T |
Early (8) |
LL |
4 |
Mild atrophy (7) |
High |
IgE-Low |
NA |
|
201531 |
V |
1 |
N, T, O |
Late (45) |
N |
No ataxia |
Normal (45) |
High |
IgA-Low |
NA |
|
201532 |
V |
3 |
N, RUL |
Late (25) |
N |
No ataxia |
Normal (~ 40) |
High |
NA |
NA |
|
N, RUL, T |
Late (30) |
N, RUL |
No ataxia |
Normal (31) |
High |
NA |
NA |
|
N, RUL |
Early (NA) |
N, RUL |
No ataxia |
NA |
High |
NA |
NA |
|
Ig: immunoglobulin; N: neck; NA: not available; T: trunk; UL: upper limb; LL: lower limb; CC: craniocervical; F: face; O: oromandibular; CR: cranial; L: limbs; H: hand; RUL: right upper limb;
G: generalized; C: classic; V: variant; MN assay: S-G2 micronucleus assay; ICBA: induced chromosome breakage assay; CSA: colony survival assay.
*prevalent ones. |
Table 4: Reported treatment of dystonia in ataxia telangiectasia patients.
|
198010 |
1 |
C |
N, T, UL |
Diazepam (high dose), haloperidol |
|
199136 |
1 |
C |
N, T, UL |
Benzhexol |
|
199416 |
1 |
C |
CC, T, UL, F |
Trihexyphenidyl |
|
200820 |
1 |
V |
N, LL |
- |
|
200921 |
1 |
V |
CC, O |
BTX |
|
201222 |
12 |
V |
N, L, CR |
BTX, anticholinergic, diazepam |
|
201325 |
3 |
V |
N |
Levodopa/carbidopa |
|
201326 |
1 |
V |
G (F, N) |
- |
|
201430 |
1 |
C |
LL,T |
Levodopa |
|
201531 |
1 |
V |
N, T, O |
Methocarbamol (slightly) |
|
201532 |
3 |
V |
N, RUL |
BTX, biperiden |
N: neck; T: trunk; UL: upper limb; LL: lower limb; CC: craniocervical; F: face; O: oromandibular; CR: cranial; L: limbs; H: hand; RUL: right upper limb;
C: classic; V: variant; BTX: botulinum toxin.
The genotype-phenotype relation in dystonia of A-T patients is not well understood. It is accepted that mutations with severe loss of ATM protein (truncating/null mutations) cause severe disease, and mild mutations (usually missense) with a residual level of protein may cause milder forms or appear late in life. In classical A-T with truncating mutation, most neurological symptoms including dystonia are similar between patients.6 However, in variant A-T, symptoms differ due to “milder” mutations, various combinations of compound heterozygous mutations, individual allelic expression pattern,32 possible modifier genes, and alternative RNA splicing.29 In addition, ATM enzymatic activity levels do not fully correlate with the milder phenotypes of variant A-T.22 Accordingly, although the genotype and phenotype comparison in A-T contributes to improved insight into the pathological mechanisms in A-T, it remains to be studied further.
Immunoglobulin class-switch recombination deficiency (CSR-D), previously known as hyper IgM syndrome, is a heterogeneous group of primary immunodeficiencies characterized by the presence of elevated or normal serum IgM levels and low or absent serum levels of the switched isotypes (IgG, IgA, and IgE). Elevated IgM is seen in 10–21% of A-T patients,37 and due to mild or delayed neurologic symptoms may be misdiagnosed with CSR-D.38 Most patients with classic A-T demonstrate immunoglobulin deficiency, but it is relatively infrequent in mild A-T forms,2,8 but may still be helpful for diagnosis.39 Based on previous publications, our patient is the first dystonic A-T with a class switching defect, causing hyper IgM immune phenotype.
Conclusion
Dystonia can be part of the clinical picture in A-T and may even mask ataxia. In clinical practice, early-onset dystonia with slow progression may be indicative of A-T. This is more important for the diagnosis of patients with the variant form of A-T, because, despite overall milder neurologic disease, the burden of malignancy remains high as for A-T, so patients will need surveillance of immunodeficiency and malignancies, as well as measures to lessen accumulating DNA damage from radiologic exposure and chemotherapeutic agents. Therefore, early-onset dystonia with cervical and brachial onset and prominent cranial involvement should be considered as a major feature of variant A-T, which may occur without general ataxia and may thus be misdiagnosed in adults with primary dystonia.
Disclosure
The authors declared no conflicts of interest.
references
- Zaki-Dizaji M, Akrami SM, Abolhassani H, Rezaei N, Aghamohammadi A. Ataxia telangiectasia syndrome: moonlighting ATM. Expert Rev Clin Immunol 2017 Dec;13(12):1155-1172.
- 2. Verhagen MM, Abdo WF, Willemsen MA, Hogervorst FB, Smeets DF, Hiel JA, et al. Clinical spectrum of ataxia-telangiectasia in adulthood. Neurology 2009 Aug;73(6):430-437.
- 3. Teive HA, Moro A, Moscovich M, Arruda WO, Munhoz RP, Raskin S, et al. Ataxia-telangiectasia - A historical review and a proposal for a new designation: ATM syndrome. J Neurol Sci 2015 Aug;355(1-2):3-6.
- 4. Taylor AM, Lam Z, Last JI, Byrd PJ. Ataxia telangiectasia: more variation at clinical and cellular levels. Clin Genet 2015 Mar;87(3):199-208.
- 5. Woods CG, Taylor AM. Ataxia telangiectasia in the British Isles: the clinical and laboratory features of 70 affected individuals. Q J Med 1992 Feb;82(298):169-179.
- 6. Nissenkorn A, Levi YB, Vilozni D, Berkun Y, Efrati O, Frydman M, et al. Neurologic presentation in children with ataxia-telangiectasia: is small head circumference a hallmark of the disease? J Pediatr 2011 Sep;159(3):466-471.
- 7. Hoche F, Frankenberg E, Rambow J, Theis M, Harding JA, Qirshi M, et al. Cognitive phenotype in ataxia-telangiectasia. Pediatr Neurol 2014 Sep;51(3):297-310.
- 8. Méneret A, Ahmar-Beaugendre Y, Rieunier G, Mahlaoui N, Gaymard B, Apartis E, et al. The pleiotropic movement disorders phenotype of adult ataxia-telangiectasia. Neurology 2014 Sep;83(12):1087-1095.
- 9. Aguilar MJ, Kamoshita S, Landing BH, Boder E, Sedgwick RP. Pathological observations in ataxia-telangiectasia. A report of five cases. J Neuropathol Exp Neurol 1968 Oct;27(4):659-676.
- 10. Bodensteiner JB, Goldblum RM, Goldman AS. Progressive dystonia masking ataxia in ataxia-telangiectasia. Arch Neurol 1980 Jul;37(7):464-465.
- 11. Byrne E, Hallpike JF, Manson JI, Sutherland GR, Thong YH. Ataxia-without-telangiectasia. Progressive multisystem degeneration with IgE deficiency and chromosomal instability. J Neurol Sci 1984 Nov-Dec;66(2-3):307-317.
- 12. Taylor AM, Flude E, Laher B, Stacey M, McKay E, Watt J, et al. Variant forms of ataxia telangiectasia. J Med Genet 1987 Nov;24(11):669-677.
- 13. Stell R, Bronstein AM, Plant GT, Harding AE. Ataxia telangiectasia: a reappraisal of the ocular motor features and their value in the diagnosis of atypical cases. Mov Disord 1989;4(4):320-329.
- 14. Friedman JH, Weitberg A. Ataxia without telangiectasia. Mov Disord 1993 Apr;8(2):223-226.
- 15. Leuzzi V, Elli R, Antonelli A, Chessa L, Cardona F, Marcucci L, et al. Neurological and cytogenetic study in early-onset ataxia-telangiectasia patients. Eur J Pediatr 1993 Jul;152(7):609-612.
- 16. Koepp M, Schelosky L, Cordes I, Cordes M, Poewe W. Dystonia in ataxia telangiectasia: report of a case with putaminal lesions and decreased striatal [123I]iodobenzamide binding. Mov Disord 1994 Jul;9(4):455-459.
- 17. Goyal V, Behari M. Dystonia as presenting manifestation of ataxia telangiectasia : a case report. Neurol India 2002 Jun;50(2):187-189.
- 18. Ball D, Rose E, Alpert E. Alpha-fetoprotein levels in normal adults. Am J Med Sci 1992 Mar;303(3):157-159.
- 19. Waldmann TA, McIntire KR. Serum-alpha-fetoprotein levels in patients with ataxia-telangiectasia. Lancet 1972 Nov;2(7787):1112-1115.
- 20. Simonin C, Devos D, Vuillaume I, de Martinville B, Sablonnière B, Destée A, et al. Attenuated presentation of ataxia-telangiectasia with familial cancer history. J Neurol 2008 Aug;255(8):1261-1263.
- 21. Carrillo F, Schneider SA, Taylor AM, Srinivasan V, Kapoor R, Bhatia KP. Prominent oromandibular dystonia and pharyngeal telangiectasia in atypical ataxia telangiectasia. Cerebellum 2009 Mar;8(1):22-27.
- 22. Saunders-Pullman R, Raymond D, Stoessl AJ, Hobson D, Nakamura K, Pullman S, et al. Variant ataxia-telangiectasia presenting as primary-appearing dystonia in Canadian Mennonites. Neurology 2012 Feb;78(9):649-657.
- 23. Nakamura K, Fike F, Haghayegh S, Saunders-Pullman R, Dawson AJ, Dörk T, et al. A-TWinnipeg: Pathogenesis of rare ATM missense mutation c.6200C>A with decreased protein expression and downstream signaling, early-onset dystonia, cancer, and life-threatening radiotoxicity. Mol Genet Genomic Med 2014 Jul;2(4):332-340.
- 24. Yanofsky RA, Seshia SS, Dawson AJ, Stobart K, Greenberg CR, Booth FA, et al. Ataxia-telangiectasia: atypical presentation and toxicity of cancer treatment. Can J Neurol Sci 2009 Jul;36(4):462-467.
- 25. Charlesworth G, Mohire MD, Schneider SA, Stamelou M, Wood NW, Bhatia KP. Ataxia telangiectasia presenting as dopa-responsive cervical dystonia. Neurology 2013 Sep;81(13):1148-1151.
- 26. Claes K, Depuydt J, Taylor AM, Last JI, Baert A, Schietecatte P, et al. Variant ataxia telangiectasia: clinical and molecular findings and evaluation of radiosensitive phenotypes in a patient and relatives. Neuromolecular Med 2013 Sep;15(3):447-457.
- 27. Cummins G, Jawad T, Taylor M, Lynch T. Myoclonic head jerks and extensor axial dystonia in the variant form of ataxia telangiectasia. Parkinsonism Relat Disord 2013 Dec;19(12):1173-1174.
- 28. Meissner WG, Fernet M, Couturier J, Hall J, Laugé A, Henry P, et al. Isolated generalized dystonia in biallelic missense mutations of the ATM gene. Mov Disord 2013 Nov;28(13):1897-1899.
- 29. Worth PF, Srinivasan V, Smith A, Last JI, Wootton LL, Biggs PM, et al. Very mild presentation in adult with classical cellular phenotype of ataxia telangiectasia. Mov Disord 2013 Apr;28(4):524-528.
- 30. Nakayama T, Sato Y, Uematsu M, Takagi M, Hasegawa S, Kumada S, et al. Myoclonic axial jerks for diagnosing atypical evolution of ataxia telangiectasia. Brain Dev 2014 Mar;37(3):362-365.
- 31. Kuhm C, Gallenmüller C, Dörk T, Menzel M, Biskup S, Klopstock T. Novel ATM mutation in a German patient presenting as generalized dystonia without classical signs of ataxia-telangiectasia. J Neurol 2015 Mar;262(3):768-770.
- 32. Lohmann E, Krüger S, Hauser AK, Hanagasi H, Guven G, Erginel-Unaltuna N, et al. Clinical variability in ataxia-telangiectasia. J Neurol 2015 Jul;262(7):1724-1727.
- 33. Liu XL, Wang T, Huang XJ, Zhou HY, Luan XH, Shen JY, et al. Novel ATM mutations with ataxia-telangiectasia. Neurosci Lett 2016 Jan;611:112-115.
- 34. Syllaba L. Contribution a I’independance de I’athetose double idiopathique’et congenitale. Atteinte faniliate, syndrome dystrophique, signe du resean vasculaire conjonctival, integrite psychique. Rev Neurol (Paris) 1926;1:541-562.
- 35. Schneider SA, Bhatia KP. Secondary dystonia–clinical clues and syndromic associations. Eur J Neurol 2010 Jul;17(Suppl 1):52-57.
- 36. Churchyard A, Stell R, Mastaglia FL. Ataxia telangiectasia presenting as an extrapyramidal movement disorder and ocular motor apraxia without overt telangiectasia. Clin Exp Neurol 1991;28:90-96.
- 37. Ghiasy S, Parvaneh L, Azizi G, Sadri G, Zaki Dizaji M, Abolhassani H, et al. The clinical significance of complete class switching defect in Ataxia telangiectasia patients. Expert Rev Clin Immunol 2017 May;13(5):499-505.
- 38. Aghamohammadi A, Imai K, Moazzami K, Abolhassani H, Tabatabaeiyan M, Parvaneh N, et al. Ataxia-telangiectasia in a patient presenting with hyper-immunoglobulin M syndrome. J Investig Allergol Clin Immunol 2010;20(5):442-445.
- 39. Motamed F, Benabbas R, Ashrafi MR, Aghamohammadi A, Rezaei N. Ataxia-telangiectasia or neurologic Wilson’s disease: when strong family history becomes a diagnostic bias. Acta Neurol Belg 2013 Jun;113(2):195-196.