Hypoparathyroidism, sensorineural deafness, and renal dysplasia (HDR) syndrome, also known as Barakat Syndrome, is a rare autosomal dominant genetic condition characterized by various degrees of renal impairment, sensorineural deafness, and hypocalcemia. Optic disc edema associated with hypoparathyroidism has been previously reported in the literature.1,2 In some cases, intracranial hypertension secondary to hypocalcemia has been identified as the cause of true papilloedema.2 However, diagnosing hypocalcemia-induced optic neuropathy can be challenging since patients do not typically present with neurological symptoms related to hypoparathyroidism.2 In this report, we present a rare case of HDR syndrome, highlighting how clues in the patient’s background history guided appropriate genetic testing and diagnosis.
Case report
A 52-year-old Caucasian man was referred to the ophthalmology department of a local hospital by his optometrist. He complained of blurred vision in his left eye, which had persisted for eight months. Recently, he also experienced discomfort behind the left eye, worsened by eye movements. Upon standing up, he reported transient vision loss in his left eye, lasting approximately five seconds, followed by an improvement. Additionally, he had headaches aggravated by bending forward or coughing, but relieved by rest or lying down. His medical history included a previous presumptive diagnosis of idiopathic intracranial hypertension, for which he had intermittently received acetazolamide. He was known to suffer loss of his right inferior visual field, which was assigned to the aforementioned, presumed neurological diagnosis. He had also been diagnosed with schizoaffective disorder and drug-induced psychosis (marijuana) and was being treated with risperidone. Hypertension was another known condition, and he used over-the-counter calcium supplements. He reported minimal alcohol consumption and was a smoker. No family history of ocular or systemic diseases was noted. He appeared relatively well during his visit to the ophthalmology clinic. His blood pressure was 173/107 mmHg, and he was apyrexial. His visual acuity was 6/5 in the right eye and 6/9 in the left eye. A relative afferent pupillary defect was observed on the right side. There was no evidence of dyschromatopsia, and his intraocular pressures were normal at 17 mmHg in both eyes. Visual field analysis revealed a stable, absolute inferior altitudinal defect in the right eye, and a relative enlargement of the blind spot in his left eye. The left optic nerve head appeared swollen, while the right nerve head displayed generalized pallor but no swelling [Figure 1]. No other abnormal neurological signs were noted during the examination. He was reported to have sensorineural hearing loss, but further details were not available at the time of presentation. Examination of the chest, cardiovascular system, and abdomen was unremarkable. The patient was referred to the medical team to investigate a possible raised intracranial pressure. Informed written consent was obtained from the patient.
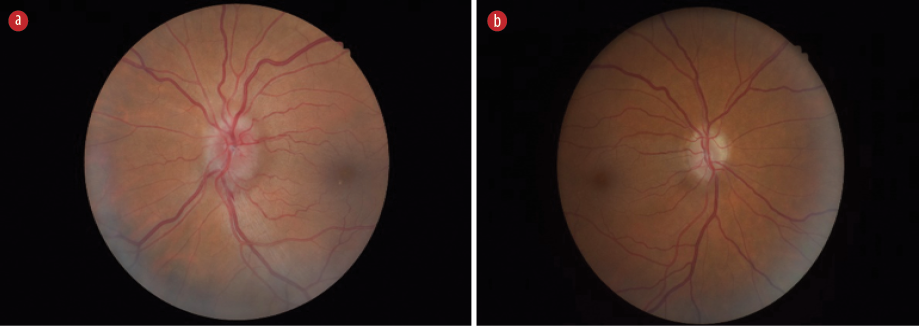 Figure 1: Optic disc appearances at presentation. (a) Left optic disc. (b) Right optic disc.
Figure 1: Optic disc appearances at presentation. (a) Left optic disc. (b) Right optic disc.
Further investigations were conducted, revealing normal results for the complete blood count. C-reactive protein was 9 mg/L with normal plasma viscosity. Urea, thyroid function, glycated hemoglobin A1c, and ferritin were all within normal limits [Table 1]. The bone profile indicated an adjusted calcium level of 1.74 mmol/L (normal range: 2.20–2.60). His phosphate level was 1.76 mmol/L (normal range: 0.80–1.50 mmol/L) with a normal alkaline phosphatase level of 100 IU/L (normal range: 30–130). Parathyroid hormone was inappropriately normal at 3.0 pmol/L (normal range: 1.6–6.9), with a low vitamin D level at 19 nmol/L (normal range: > 51). A head computed tomography scan did not reveal any compatible features suggestive of raised intracranial pressure. A lumbar puncture revealed an opening pressure of 22 cm and a closing pressure of 18 cm of cerebral spinal fluid with normal constituents.
Table 1: Blood results on initial presentation.
|
ALP, IU/L
|
100
|
30–130
|
|
ALT, U/L
|
12
|
0–45
|
|
Bilirubin, umol/L
|
4
|
< 21
|
|
K+, mmol/L
|
4.5
|
3.5–5.3
|
|
Creatinine, umol/L
|
110
|
59–104
|
|
Urea, mmol/L
|
5.5
|
2.5–7.8
|
|
CRP, mg/L
|
9
|
0–5
|
|
Plasma viscosity, mPa.s
|
1.58
|
1.50–1.72
|
|
Phosphate, mmol/L
|
1.76
|
0.80–1.50
|
|
Vitamin D, nmol/L
|
19
|
> 51
|
|
HbA1C, mmol/mol
|
38
|
20–41
|
|
TSH, mU/L
|
0.8
|
0.27–4.20
|
|
Free T4, pmol/L
|
22
|
11–22
|
ALP: alkaline phosphatase; ALT: alanine amino transferase; K: potassium; CRP: c-reactive protein; HbA1c: hemoglobin A1c; TSH: thyroid
stimulating hormon.
The case was discussed with the endocrine team, and a diagnosis of hypocalcemia secondary to hypoparathyroidism was established. Advice was provided over the phone, and an outpatient appointment was scheduled. In a subsequent review by the ophthalmology team, a diagnosis of left optic nerve head swelling and previous right optic atrophy due to hypercalcemic optic neuropathy was made. The right optic atrophy was attributed to previous ischemia caused by optic disc edema. The patient was managed as an outpatient and prescribed alfacalcidol (1 microgram once a day) along with oral calcium supplements. The patient attended a follow-up visit in the endocrine clinic, where a detailed medical and family history was taken. This revealed that he had sensorineural deafness. His son had received treatment for hypoparathyroidism and deafness, while his daughter had been diagnosed with end-stage kidney disease secondary to renal dysplasia in addition to hypocalcemia (although she was never formally referred to endocrinology). Additionally, she underwent bilateral cataract extraction when she was 10 years old, despite the absence of a family history of cataracts. The persistent hypocalcemia was considered the most likely cause of her ocular presentation.
A renal ultrasound revealed a hypoplastic left kidney, but analysis of the patient’s renal function was unremarkable. Genetic testing confirmed that the patient was heterozygous for a pathogenic GATA3 variant, confirming the diagnosis of autosomal dominant HDR syndrome. His family members were informed and referred to a clinical genetics service. During the follow-up, his calcium levels were measured and improvement was shown [Table 2]. He also reported a significant improvement in cognition and overall well-being.
Table 2: Adjusted calcium levels during follow-up.
|
23.12.2021 (Baseline)
|
1.74
|
|
24.12.2021
|
2.09
|
|
06.01.2022
|
1.91
|
|
17.01.2022
|
1.91
|
|
24.01.2022
|
2.02
|
|
15.02.2022
|
2.17
|
He was reviewed in the eye clinic three weeks after his initial assessment. His visual acuity improved to 6/6 in the right eye and remained 6/9 in the left eye. His left optic nerve head showed a reduction in swelling. The post-treatment optic disc appearances are shown in Figure 2. At the latest review, his calcium level was 2.41. The left optic nerve head swelling had resolved, and the right optic disc displayed mild general optic disc pallor (from previous ischemic optic neuropathy) without swelling. His final visual acuities were 6/4 in the right eye and 6/7.5 in the left eye, demonstrating improvement compared to the initial presentation at the ophthalmology clinic. Visual field assessment revealed a stable inferior absolute altitudinal defect in the right eye and a full visual field in the left eye [Figure 3].
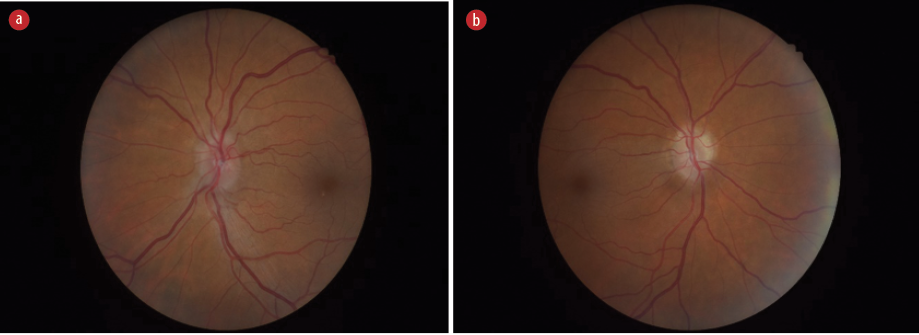 Figure 2: Post-treatment optic discs following hypocalcemia treatment. (a) Left optic disc. (b) Right
Figure 2: Post-treatment optic discs following hypocalcemia treatment. (a) Left optic disc. (b) Right
optic disc.
 Figure 3: Visual fields post-treatment showing a stable inferior absolute altitudinal defect in the right eye and a full visual field in the left eye.
Figure 3: Visual fields post-treatment showing a stable inferior absolute altitudinal defect in the right eye and a full visual field in the left eye.
Discussion
Our patient presented with hypoparathyroidism and deafness without renal impairment. Approximately 28.6% of patients with HDR syndrome have only hypoparathyroidism and deafness without renal involvement.3 Hypoparathyroidism in HDR syndrome leads to hypocalcemia, which can be asymptomatic or present with muscle aches/spasms, neuromuscular irritability, seizures, or tetany.
Hypocalcemia is a rare but well-documented cause of optic disc edema. Patients with hypocalcemia-induced optic disc edema are uncommonly presented to the eye clinic, which can contribute to delays in diagnosis. To the best of our knowledge, this may be the first reported case of HDR syndrome diagnosed following presentation with hypocalcemia-induced optic disc edema.
Disc swelling in patients with hypocalcemia can result from increased intracranial pressure (papilledema) or disruption of axoplasmic flow due to low calcium levels in patients without evidence of intracranial hypertension.1 Low calcium levels can lead to hypersecretion of cerebrospinal fluid, resulting in the former neuro-ophthalmic presentation, while the latter presentation may lead to ischemic optic neuropathy. Therefore, clinicians need to be aware of this rare cause of optic disc edema and assess serum calcium levels when referring patients to the medical team for further neurological investigations. Although our patient's diagnosis was made relatively quickly once referred by the ophthalmology team, a case report highlighted the risk of performing expensive and invasive investigations if serum calcium levels were not checked earlier in the diagnostic workup.1 In fact, our patient had been previously reviewed and treated for raised intracranial pressure, despite normal opening cerebrospinal fluid opening pressures, which in retrospect may have been due to hypocalcemia-induced optic disc edema.
HDR syndrome has been shown to have genetic heterogeneity.4 In our patient, genetic studies confirmed a GATA3 gene mutation, while deletions in chromosome 10p14 can also cause the syndrome. Given the presence of hypoparathyroidism-induced hypocalcemia in our patient, it was essential to consider a genetic cause. Upon reviewing previous pathology results, there was evidence of persistently low calcium levels. Combined with a history of deafness and a family history of hypocalcemia and renal impairment, these findings raised suspicions of an underlying syndrome.
The genetic diagnosis has led to the diagnosis of HDR in the aforementioned family members.
Conclusion
Hypocalcemia-induced disc edema is a rare but possible presentation of HDR syndrome. Patients presenting with features of optic nerve disc edema should have their serum calcium levels measured. Additionally, patients with hypoparathyroidism-associated hypocalcemia should have an evaluation of their personal and family history, specifically exploring the presence of deafness and renal dysfunction, as this may lead to the diagnosis of HDR syndrome.
Disclosure
The authors declared no conflicts of interest.
references
- 1. Abu-Ain M, Aazem S, Morton C, Kumwenda M, Griffiths D, Jacob A. A rare potentially treatable cause of bilateral optic disc swelling. BMJ Case Rep 2010 Oct;2010:bcr0320102835.
- 2. Sforza G, Deodati A, Moavero R, Papetti L, Frattale I, Vigevano F, et al. Benign intracranial hypertension due to hypoparathyroidism: a case report. Front Neurol 2022 Jan;12:818638.
- 3. Joseph AD, Sirisena ND, Kumanan T, Sujanitha V, Strelow V, Yamamoto R, et al. Hypoparathyroidism, Sensorineural deafness and renal disease (Barakat syndrome) caused by a reduced gene dosage in GATA3: a case report and review of literature. BMC Endocr Disord 2019 Oct;19(1):111.
- 4. Barakat AJ, Raygada M, Rennert OM. Barakat syndrome revisited. Am J Med Genet A 2018 Jun;176(6):1341-1348.