| |
Abstract
This is the first report of congenital adrenal hyperplasia (CAH) due to combined 17α-hydroxylase/17,20 lyase deficiency in an Omani patient who was initially treated for many years as a case of hypertension. CAH is an uncommon disorder that results from a defect in steroid hormones biosynthesis in the adrenal cortex. The clinical presentation depends on the site of enzymatic mutations and the types of accumulated steroid precursors. A 22-year-old woman who was diagnosed to have hypertension since the age of 10 years who was treated with anti-hypertensive therapy was referred to the National Diabetes and Endocrine Centre, Royal Hospital, Oman. The patient also had primary amenorrhea and features of sexual infantilism. Full laboratory and radio-imaging investigations were done. Adrenal steroids, pituitary function and karyotyping study were performed and the diagnosis was confirmed by molecular mutation study. Laboratory investigations revealed adrenal steroids and pituitary hormones profile in addition to 46XY karyotype that are consistent with the diagnosis of CAH due to 17α-hydroxylase deficiency. Extensive laboratory workup revealed low levels of serum cortisol (and its precursors 17α-hydroxyprogesterone and 11-deoxycortisol), adrenal androgens (dehydroepiandrosterone sulfate and androstenedione), and estrogen (estradiol); and high levels of mineralocorticoids precursors (11-deoxycorticosterone and corticosterone) with high levels of ACTH, FSH and LH. Mutation analysis revealed CYP17A1-homozygous mutation (c.287G>A p.Arg96Gln) resulting in the complete absence of 17α-hydroxylase/17,20-lyase activity. The patient was treated with dexamethasone and ethinyl estradiol with cessation of anti-hypertensive therapy. A review of the literature was conducted to identify previous studies related to this subtype of CAH. This is the first biochemically and genetically proven case of CAH due to 17α-hydroxylase/17,20-lyase deficiency in Oman and in the Arab World described in the literature.
Keywords: Congenital adrenal hyperplasia; 17 α-hydroxylase; 17,20-lyase; Hypertension; Pseudohermaphroditism; Adrenal cortex; Oman.
Introduction
Hypertension is uncommon during childhood and adolescence with its presence usually pointing to an underlying etiology mostly of metabolic origin. Of particular importance in its causations especially when associated with genital ambiguity or disordered sex development are mutations in the adrenal steroidogenesis mostly due to ‘congenital adrenal hyperplasia’ (CAH). The deficiency forms of CAH which are associated with male pseudohermaphrodite include 21α-hydroxylase and to a lesser extent 11β-hydroxylase; with female pseudohermaphrodite include 17α-hydroxylase, 17,20 lyase, 3β-hydroxysteroid dehydrogenase, and cholesterol 20,22 desmolase; while CAH forms which are associated with hypertension include 17α-hydroxylase and 11β-hydroxylase deficiency.1 Other inherited metabolic disorders that may be associated with monogenic hypertension during childhood and adolescence include apparent mineralocorticoid excess, glucocorticoid remediable aldosteronism, familial hyperaldosteronism type 2, Liddle's syndrome, Gordon's syndrome, activating mutations of the mineralocorticoid receptor, and generalized glucocorticoid resistance.2
Steroidogenesis is an essential pathway for the synthesis of steroid hormones in the adrenal cortex: mineralocorticoids, glucocorticoids and androgens, mainly dehydroepiandrosterone (DHEA), and androstenedione. Cholesterol is the key substrate in this pathway which is converted by the rate limiting cholesterol side chain cleavage enzyme (CYP11A1) into pregnenolone, the common precursor for all steroid hormones. In the adrenal cortex, this is processed into either glucocorticoids and androgens in zona fasciculate and zona reticularis (which contain the microsomal enzyme cytochrome P450c17α-hydroxylase: P450c17 and 17,20 lyase) or into mineralocorticoids in zona glomerulosa (which does not contain P450c17 and 17,20 lyase, but contains aldosterone synthase, CYP11B2). Because it possesses both 17α-hydroxylase and 17,20 lyase activities, P450c17 is considered a bifunctional enzyme that has a profound impact on the flux through the steroidogenic pathway for its selective cellular localization in zona fasciculate and zona reticularis and its absence in zona glomerulosa.1,3 This enzyme is encoded by the CYP17A1 gene and has been mapped to chromosome 10q24-25 in the adrenal cortex (zona reticularis) and gonads.4 The protein is localized in the endoplasmic reticulum, has both 17α-hydroxylase and 17,20 lyase activities, and is considered as the key enzyme in steroidogenesis for producing: progestins, mineralocorticoids, glucocorticoids, androgens and estrogens. Specifically, CYP17A1 acts upon pregnenolone and progesterone to add a hydroxyl group (-OH) at carbon 17 position of the steroid D ring, or acts upon 17α-hydroxypregnenolone and 17α-hydroxprogesterone to split the nucleus (17,20 lyase activity) to convert them into C-19 androgen precursors, DHEA and androstenedione, respectively. For understanding the biosynthesis of adrenals steroids, a scheme of the pathway is presented. (Fig 1)
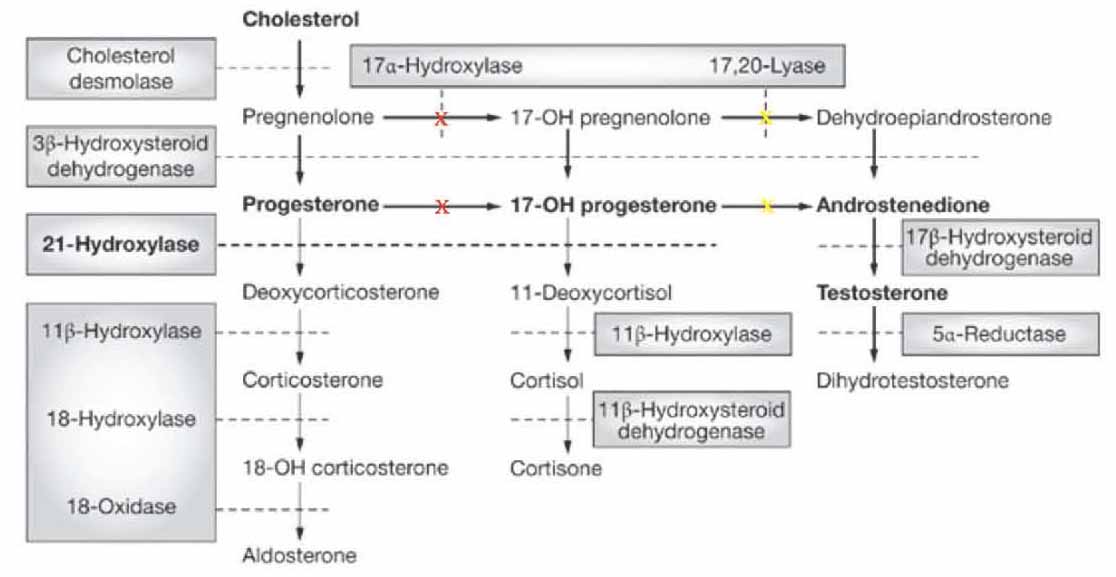
Figure 1: Major human steroidogenic pathway in the adrenal cortex. Key enzymes are shown as dashed arrows/shaded boxes indicating the chemical reactions. P450scc (cholesterol desmolase) cleaves cholesterol to pregnenolone, the first intermediate in steroid biosynthesis. The steroids in the first row are Δ5-steroids, which constitute the preferred pathway to C19 steroids in human. Not all intermediate steroids, pathways, and enzymes are shown. In CAH due to 17α-hydroxylase deficiency (marked as red X) there is overproduction of the precursors/hormones on the left side of the first column with decreased production of the precursors/hormones on the right side of the second and third columns. 'Adopted from: Nimkarn S, New M. Prenatal diagnosis and treatment of congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Nat Clin Pract Endocrinol Metab 2007 May;3(5):405-413'.
In CAH, there are various genetic mutations in the enzymes involved in steroidogenesis. Due to this enzymatic defect, cortisol is under-produced and the negative feedback control on ACTH is lost with consequently excess ACTH produced in order to normalize cortisol levels resulting in overproduction and accumulation of steroids precursors prior to the enzyme defect as well as hyperplasia of the adrenal cortex. The clinical manifestation depends on the level of enzyme block in the steroid synthesis. CAH should be considered in infants, children or adolescents with ambiguous genitalia, sexual infantilism, hypogonadism or hypertension, particularly when associated with disturbed water, electrolytes and hydrogen homeostasis. The most common form is 21α-hydroxylase deficiency which may be diagnosed at birth by the presence of virilization in female infants or by features of salt wasting in both genders.5 The 11β-hydroxylase deficiency is uncommon and 17α-hydroxylase deficiency is a rare form of CAH which may present much later in adolescence or adulthood.6,7
Case Report
A 22-year-old Omani woman was referred from Nizwa Hospital, a secondary care regional hospital, to the National Diabetes and Endocrine Centre, Royal Hospital, a tertiary care hospital in Oman, for expert consultation. She was diagnosed to have hypertension at the age of 10 years and was considered to have early essential hypertension for which she was given antihypertensive therapy. At the time of her consultation at Royal Hospital, she was on three antihypertensive medications: β1 receptor antagonist (atenolol), angiotensin-converting enzyme inhibitor (lisinopril) and diuretic (thiazide). However, despite this combined therapy, her hypertension was not well controlled. The patient also had primary amenorrhea and features of sexual infantilism and she appeared to have pseudohermaphroditism. She had no past history of hernial surgery. The family history was unremarkable. She has 8 siblings (2 brothers and 6 sisters).
On physical examination, she was quite tall with height 173 cm, weight 73 kg, BMI 24.2 kg/m2, BP 145/90 mmHg, with no secondary sexual characteristics, no pubic or axillary hair, Tanner score P1B1, external genitalia of female type, with no goiter. Diagnostic laparoscopy revealed no internal female organs, ovaries or uterus. No clinical signs or stigmata of familial hypercholesterolemia were noticed. On radio-imaging studies, both ultrasound examination of the abdomen and pelvis, and MRI of the abdomen and pelvis showed no uterus, ovaries or testicles. There was some fibrous tissue detected in the vagina.
Laboratory investigations revealed results within the reference ranges for core blood laboratory tests including electrolytes, renal, liver, bone, glucose, HbA1c and thyroid profiles, with moderate hypercholesterolemia. Concerning endocrine tests, the results of blood profile for the adrenal and gonadal steroids, pituitary hormones and karyotype study are shown in Table 1. She had high FSH and LH with low serum estradiol (E2), consistent with primary gonadal failure (hypergonadotrophic hypogonadism). She also had low cortisol (and its precursors 17α-hydroxyprogesterone and 11-deoxycortisol), low adrenal androgens (DHEA sulfate and androstenedione), high mineralocorticoids precursors (11-deoxycorticosterone and corticosterone), high ACTH, and chromosomal analysis revealed a normal male karyotype 46 XY. All these clinical, imaging and laboratory data are consistent with CAH due to 17α-hydroxylase deficiency.
Table 1: Plasma steroids and pituitary hormones at time of presentation using different reporting units (compared with recommended adult reference ranges).
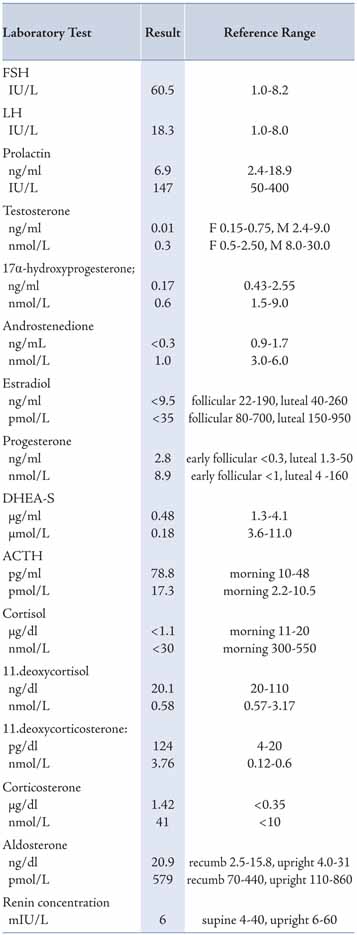
All core clinical biochemistry and hormonal tests were performed at Royal Hospital, Oman, using Architect 8000c and Architect 2000i, respectively, with Automated Processing System (Abbott, USA); and 17α-hydroxyprogesterone, renin and aldosterone were performed by ELISA (DRG, USA); while 11-deoxycortisol, 11-deoxycorticosterone, corticosterone, androstenedione and ACTH were performed at Laboratoroire Cerba (France).
For molecular testing, request with patient’s consent for CYP17A1 gene analysis (17α-hydroxylase deficiency, OMIM 202110; inheritance: autosomal recessive) was sent to Centogene Laboratory (Centrogene AG, Schillingallee 68, 18057 Rostock, Germany). The CYP17A1 gene was analyzed by PCR and sequencing of both DNA strands of the entire coding region and the highly conserved exon-intron splice junctions. The reference sequence of the CYP17A1 gene is: NM_000102.3. The results revealed: CYP17A1 - homozygous mutation (c.287G>A p.Arg96Gln) which is compatible with a diagnosis of CAH due to 17α-hydroxylase deficiency. A homozygous mutation in exon 1 of the CYP17A1 gene (c.287G>A p.Arg96Gln) was found. This mutation has previously been described as disease-causing by Brooke et al.8 in 2006 (Human Gene Mutation Database; HGMD Professional 2013.2) with this mutation affecting an amino acid in a key substrate-binding region of the enzyme, leading to complete inactivity. Genetic counseling was recommended and the patient’s family was informed.
As with other CAH variants, appropriate glucocorticoid replacement is the cornerstone of therapy. Accordingly, once the diagnosis was made, the patient was started and maintained on steroid therapy, dexamethasone 0.5 mg once daily, with estrogen replacement, ethinyl estradiol 2 µg once daily to improve the female external appearance as the patient did not consider any change in her sex from female phenotype.
The patient was asked to withdraw the antihypertensive drugs one by one with blood pressure monitoring. The patient was also advised lifestyle changes and was given lipid-lowering therapy, atorvastatin 10 mg once daily. Three months after the diagnosis, the laboratory investigations revealed normal levels of ACTH 6.0 pg/mL and corticosterone 0.165 µg/dL, with BP stable at 130/80 mmHg. The patient was maintained and continued on dexamethasone 0.5 mg once daily and ethinyl estradiol 2 µg once daily.
Discussion
This report describes a biochemically and genetically proven case of combined 17α-hydroxylase/17,20 lyase deficiency that is first to be reported in Oman. The 22-year-old patient had this metabolic disorder masquerading as hypertension since childhood with sexual infantilism and primary amenorrhea. The clinical, imaging and laboratory findings (adrenal steroids, pituitary hormones, and karyotyping) are characteristic of CAH due to 17α-hydroxylase deficiency.6 The diagnosis was confirmed by mutation analysis which revealed a novel homozygous missense mutation in CYP1 17A1 affecting an amino acid in a key substrate binding region of the enzyme (homozygous mutation in exon 1 of the CYP17A1 gene; c.287G>A p.Arg96Gln) resulting in the complete absence of 17α-hydroxylase/17,20 lyase activity. This rare mutation has been described by Brooke et al.8 in 2006 in a 17-year-old female presenting with a malignant mixed germ cell tumor with clinical and biochemical features of combined 17α-hydroxylase/17,20 lyase deficiency. Mutations in CYP17A1 gene were first reported by Kagimoto et al.9 in 1988. Now more than 50 different mutations in exons and introns of this gene have been reported to cause complete or partial 17α-hydroxylase deficiency.6,10,11
This CAH variant is a rare autosomal recessive disorder that constitutes approximately 1% of all CAH cases.4 It was first described in 1966 by Biglieri et al.12 in a 35-year-old phenotypic female with hypertension, sexual infantilism and primary amenorrhea. The report was followed by other reports by Goldsmith et al.13 in 1967, Mallin in 1969,14 and New in 1979.15 Now, there are more than 150 cases that are reported in literature, the latest by Lee et al.16 in 2013. As per literature search in Pubmed in December 2013, the current case report appears to be the first biochemically and genetically proven case of CAH due to 17α-hydroxylase/17,20 lyase deficiency caused by CYP17A1 gene mutation diagnosed and reported in Oman, as well as in the Arab World.
Deficiency in CYP17A1 results in decreased synthesis of glucocorticoids and androgens (DHEA and androstenedione). Accordingly, pregnenolone will not be 17α-hydroxylated to 17α-hydroxypregnenolone leading to its conversion through 3β-hydroxysteroid dehydrogenase pathway into progesterone which also in turn will not be 17α-hydroxylated to 17α-hydroxyprogesterone. Accumulation of these substrates, pregnenolone and progesterone will enhance the 21-hydroxylation and 11β-hydroxylation steps in the mineralocorticoid pathway resulting in an overproduction of 11-deoxycorticosterone and corticosterone. Deficiency in 17α-hydroxylase will lead to a decrease in all the subsequent steroids namely: 17α-hydroxypregnenolone, 17α-hydroxylprogesterone, DHEA, androstenedione, testosterone, 11-deoxycortisol and cortisol. The resultant decline in cortisol and its feedback inhibition on pituitary ACTH will lead to an increase in ACTH release in order to bring cortisol production to normal levels and so leading to overstimulation of the steroid synthetic pathway at the expense of excess formation and accumulation of steroid precursors prior to the enzymatic block of 17α-hydroxylase and their products, particularly mineralocorticoids precursors (11-deoxycorticosterone and corticosterone), as well as hyperplasia of the adrenal cortex.1,2 For genital organs development, the two enzymes, 17α-hydroxylase/17,20 lyase and 3β-hydroxysteroid dehydrogenase, in both the adrenals and gonads are shared in sexual maturity throughout fetal life and puberty. Thus in CAH due to 17α-hydroxylase deficiency, there will be an impairment of both adrenal and gonadal steroid hormones (androgens and estrogens). Male patients will have gonads but with no internal male genitalia such as prostate, seminal vesicles and vas deference, because of the lack of testosterone that is required for Wolffian duct development. The anti-Mullierian hormone AMH (also known Mullerian inhibiting factor, MIF; or Mullerian-inhibiting hormone, MIH) is still present from the sertoli cells.
In our case, biochemically there were decreased concentrations in: 17α-hydroxy progestrone, androstenedione, testosterone, estradiol, 11-deoxycortisol and cortisol; with increased concentrations in: progesterone, 11-deoxycorticosterone and corticosterone, as well as ACTH. The steroid profile was consistent with CAH due to 17α-hydroxylase deficiency. The decreased production of sex steroids causes under-virilization of the external genitalia with disordered sex development (no pubertal features) and primary amenorrhea in this 46XY patient. The raised levels of gonadotrophins (FSH and LH) indicate primary gonadal failure. The low cortisol and high ACTH levels reflect a state of primary adrenal hypocortisolism; however, adrenal insufficiency was not manifesting itself as a classical Addison's disease as the raised corticosterone production has weaker glucocorticoid effect. Also, the high 11-deoxycorticosterone has potent mineralocorticoid effect that causes sodium retention and potassium/hydrogen loss and consequently hypertension, which is present in most patients although 10% to 15% may be normotensive at the time of diagnosis.6,17 This may result in the inhibition of renin-angiotensin system and decreased aldosterone synthesis as hyporeninemic hypoaldosteronism or hyporeninemic normoaldosteronism. In our patient, the antihypertensive therapy (atenolol, lisinopril and thiazide) might have also had an affect on sodium, potassium and hydrogen ion handling in the kidney and have influenced the electrolytes pattern.
As with other CAH variants, appropriate glucocorticoid replacement is the cornerstone of therapy with the aim to decrease ACTH and 11-deoxycorticosterone to normal using the smallest dose, dexamethasone (0.25-1.0 mg/d) or prednisone (2-5 mg/d). Appropriate glucocorticoid replacement usually normalizes the blood pressure, and serum renin, aldosterone and electrolytes levels with natriuresis. Hormone replacement regimens have to start early in adolescence to allow development of secondary sexual characteristics and stimulate normal anabolic bone mass of puberty. The 46,XX patients usually require combined/cyclic estrogen/progestin therapy to prevent endometrial hyperplasia from unopposed estrogen and are often treated with estrogen alone if they first come to medical attention at the time of puberty, or at diagnosis if that time has passed and with the decision to be reared as females. In case the patients decide to be males, then they should be treated with androgen replacement with consideration of extensive genital reconstructive surgery including gonadectomy to avoid malignant degeneration in the intra-abdominal testes. This treatment should be planned following education of the patient and parents regarding its complexity, implications and consequences.10 Our patient was maintained on dexamethasone 0.5 mg once daily and ethinyl estradiol 2 µg once daily for the secondary sexual features as the patient did not consider any change in her sex.
Conclusion
We have reported the first biochemically and genetically proven case of CAH due to 17α-hydroxylase/17,20 lyase deficiency in Oman and in the Arab World. The diagnosis of this subtype of CAH may be delayed and may present with hypertension in late childhood, adolescence or adulthood with improper development of sexual milestones and characteristics. A high index of clinical suspicion is necessary when evaluating children and adolescents with hypertension, particularly in the presence of superadded sexual infantilism with ambiguous external genitalia. The diagnosis is usually made by finding the characteristic pattern in the adrenal steroid profile with confirmation by molecular mutation analysis.
Acknowledgements
The authors would like to thank Waleed Pharmacy, Muscat, Oman, for their support in sponsoring the molecular testing for this patient which was performed at Centogene Laboratory (Centrogene AG, Schillingallee 68, 18057 Rostock, Germany). No conflict of interests to declare.
References
1. Honour JW. Diagnosis of diseases of steroid hormone production, metabolism and action. J Clin Res Pediatr Endocrinol 2009;1(5):209-226.
2. Hassan-Smith Z, Stewart PM. Inherited forms of mineralocorticoid hypertension. Curr Opin Endocrinol Diabetes Obes 2011 Jun;18(3):177-185.
3. Van Den Akker EL, Koper JW, Boehmer AL, Themmen AP, Verhoef-Post M, Timmerman MA, et al. Differential inhibition of 17alpha-hydroxylase and 17,20-lyase activities by three novel missense CYP17 mutations identified in patients with P450c17 deficiency. J Clin Endocrinol Metab 2002 Dec;87(12):5714-5721.
4. Matteson KJ, Picado-Leonard J, Chung BC, Mohandas TK, Miller WL. Assignment of the gene for adrenal P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase) to human chromosome 10. J Clin Endocrinol Metab 1986 Sep;63(3):789-791.
5. Krone N, Arlt W. Genetics of congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab 2009 Apr;23(2):181-192.
6. Karter CE, Biglieri EG. Disorders of steroid 17 α-hydroxylase deficiency. Endocrinol Metab North Am 1994 Jun;23(2):341-357.
7. Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev 2011 Feb;32(1):81-151.
8. Brooke AM, Taylor NF, Shepherd JH, Gore ME, Ahmad T, Lin L, et al. A novel point mutation in P450c17 (CYP17) causing combined 17alpha-hydroxylase/17,20-lyase deficiency. J Clin Endocrinol Metab 2006 Jun;91(6):2428-2431.
9. Kagimoto M, Winter JS, Kagimoto K, Simpson ER, Waterman MR. Structural characterization of normal and mutant human steroid 17 α-hydroxylase genes: molecular basis of one example of combined 17 α-hydroxylase/17,20 lyase deficiency. Mol Endocrinol 1988 Jun;2(6):564-570.
10. Auchus RJ. The genetics, pathophysiology, and management of human deficiencies of P450c17. Endocrinol Metab Clin North Am 2001 Mar;30(1):101-119, vii. vii.
11. Yao F, Huang S, Kang X, Zhang W, Wang P, Tian Q. CYP17A1 mutations identified in 17 Chinese patients with 17α-hydroxylase/17,20-lyase deficiency. Gynecol Endocrinol 2013 Jan;29(1):10-15.
12. Biglieri EG, Herron MA, Brust N. 17-hydroxylation deficiency in man. J Clin Invest 1966 Dec;45(12):1946-1954.
13. Goldsmith O, Solomon DH, Horton R. Hypogonadism and mineralocorticoid excess. The 17-hydroxylase deficiency syndrome. N Engl J Med 1967 Sep;277(13):673-677.
14. Mallin SR. Congenital adrenal hyperplasia secondary to 17-hydroxylase deficiency. Two sisters with amenorrhea, hypokalemia, hypertension, and cystic ovaries. Ann Intern Med 1969 Jan;70(1):69-75.
15. New MI. Male pseudohermaphroditism due to 17 alpha-hydroxylase deficiency. J Clin Invest 1970 Oct;49(10):1930-1941.
16. Lee ES, Kim M, Moon S, Jekarl DW, Lee S, Kim Y, et al. A new compound heterozygous mutation in the CYP17A1 gene in a female with 17α-hydroxylase/17,20-lyase deficiency. Gynecol Endocrinol 2013 Jul;29(7):720-723.
17. Yanase T, Simpson ER, Waterman MR. 17 α-hydroxylase/17,20-lyase deficiency: from clinical investigation to molecular definition. Endocr Rev 1991 Feb;12(1):91-108.
|