| |
Abstract
Rasmussen encephalitis is an extremely rare chronic inflammatory neurodegenerative disease affecting a single cerebral hemisphere, causing progressive neurological deterioration and intractable seizures. Imaging plays an important role in diagnosis by demonstrating focal or unihemispheric involvement and excluding other possible causes. Here, we report a case of Rasmussen encephalitis with an update on recent diagnostic criteria and emphasis on differential diagnoses which can be excluded on imaging.
Keywords: Rasmussen encephalitis; Magnetic resonance imaging; Epilepsia partialis continua.
Introduction
Rasmussen encephalitis (RE) is a rare disease of unknown etiology that causes severe chronic unihemispheric inflammatory disease of the central nervous system mainly in children. It leads to intractable seizures, cognitive decline and progressive neurological deficits in the affected hemisphere.1 Though the etiology of this sporadic disease is unclear, cytotoxic T cell reaction against the neurons was implicated in the pathogenesis.2 Imaging plays a pivotal role in diagnosis by exclusion of other causes and helps towards monitoring the disease progress. Early institution of immunotherapy has been suggested to improve the outcome and alter the natural history of disease.3 Here, we report of a young girl diagnosed with RE based on clinical features, electroencephalography (EEG) and imaging findings.
Case Report
An 8-year-old girl presented to the Neurology clinic with clonic movements of the right hand and leg for several months. Later on, they progressed to continuous partial seizures of the right leg associated with difficulty in walking. For the past one year, she was having mild orbito-frontal headache and decreased vision bilaterally. Her perinatal period and developmental milestones had been normal. On examination, visual acuity in both eyes was 6/24 and the right lower limb showed decreased tone and power (3/5).
Routine blood and cerebrospinal fluid investigations and metabolic tests were within normal limits except for positive antinuclear antibody (ANA) screening. The rest of ANA profile was normal. EEG showed epilepsia partialis continua with electrographic correlation. Magnetic resonance imaging (MRI) showed focal hyperintensity in the left superior frontal gyrus on T2-weighted (T2W) and fluid-attenuated inversion recovery (FLAIR) images (Fig. 1). Atrophy of the left cerebral hemisphere was noted evidenced by dilatation of the ipsilateral lateral ventricle and widening of the cortical sulci, most marked at the temporal lobe (Fig. 2). To exclude a vasculitic etiology, a digital subtraction angiography (DSA) was performed which was normal. Paraneoplastic cause was excluded by a normal computed tomography (CT) scan of the chest and abdomen. Considering all the above factors, diagnosis of Rasmussen encephalitis was suggested.
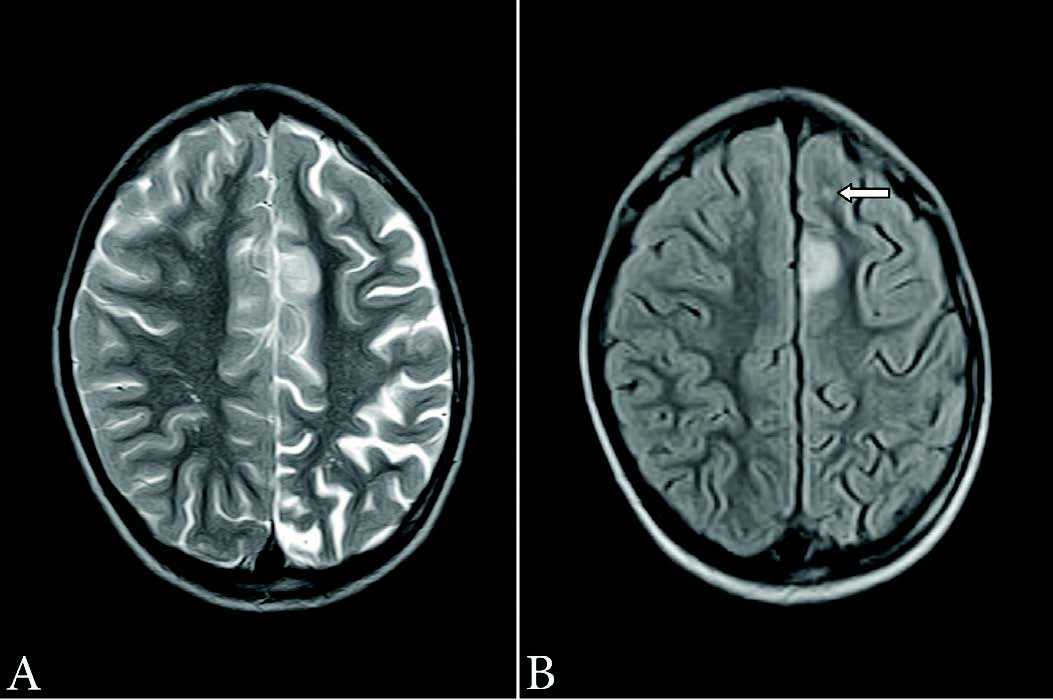
Figure 1: (a) Axial T2W image (TR: 4224 ms, TE: 99 ms, slice thickness: 4 mm) showing an area of hyperintense signal in left superior frontal region along with widening of cortical sulci on left side; (b) Axial FLAIR image (TR: 7800 ms, TE: 110 ms, slice thickness: 4 mm) showing hyperintense signal in left superior frontal cortex. A small hyperintense focus is also seen in anterior white matter (arrow).
The patient was initially treated with antiepileptic medication. Treatment with intravenous gamma globulin and prednisolone was started later based on the diagnosis of RE. Motor function of the right leg improved mildly. Partial control of the seizures was attained. The clinical condition remained almost static with medication on follow-up for seven months.
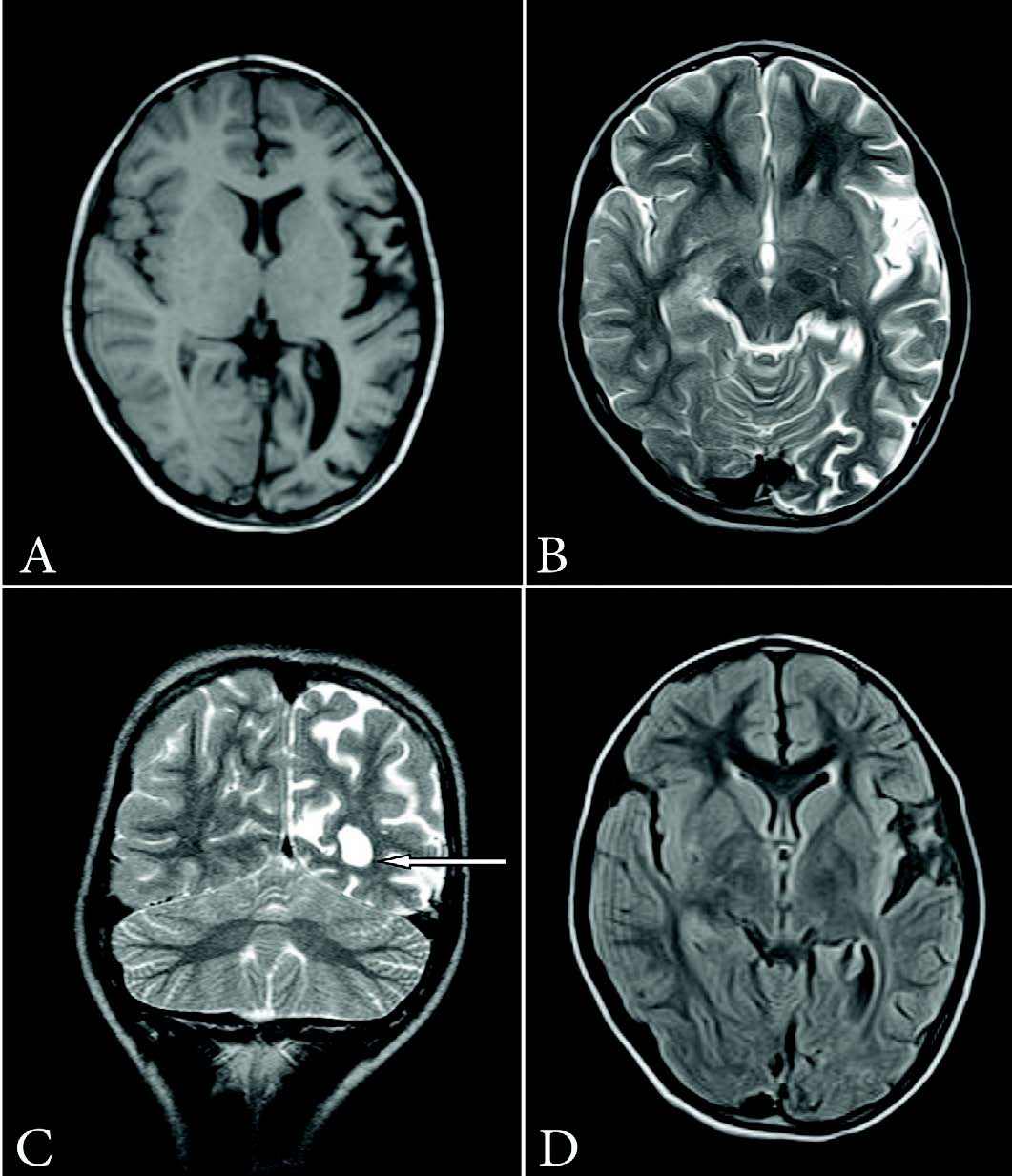
Figure 2: (a) Axial T1-weighted image (TR: 660 ms, TE: 14 ms, slice thickness: 4 mm) showing atrophy of left hemisphere; (b)Axial T2W image (TR: 4224 ms, TE: 99 ms, slice thickness: 4 mm) showing widening of cortical sulci and sylvian fissure on the left side; (c) Coronal T2-weighted image (TR: 5979 ms, TE: 99 ms, slice thickness: 3 mm) demonstrating dilatation of left lateral ventricle (arrow) and widening of cortical sulci; (d) Axial FLAIR image (TR: 7800 ms, TE: 110 ms, slice thickness: 4 mm) showing features of volume loss on the left side.
Discussion
RE is a sporadic chronic inflammatory disease of the central nervous system occurring mostly in the pediatric population, first reported by Theodore Rasmussen in 1958. The mean age of presentation is between 6 to 8 years. Both sexes are equally affected.1 Our patient was in the same age group. The etiology of RE is unknown, with some earlier studies suggesting the role of viral infections, while others describing it as an autoimmune phenomenon involving antibodies against a protein of glutamate receptor.3,4 According to a recent concept, cytotoxic T cell reaction against the neuron leads to expression of major histocompatibility complex (MHC) class I and apoptotic neuronal death, resulting in progressive deterioration of neurological status.2 No specific etiology could be found in our patient either. Clinically, RE presents as epilepsia partialis continua (EPC) followed by hemiparesis and cognitive impairment, which gradually progresses with the disease activity.
Diagnosis of RE is based on characteristic clinical, radiological, and pathological features with more emphasis on clinico-radiological features, as brain biopsy, due to its invasive nature, is not done in all the cases. Although the reported cohorts are not large, Bien et al. proposed a three stage natural history of RE on the basis of long term observation of 13 patients.5,6 The first stage is nonspecific, called the prodromal stage, manifesting with a relatively low seizure frequency and, rarely, mild hemiparesis (median duration: 7.1 months; range: 0 months to 8.1 years). The second phase, called the acute stage with a median duration of 8 months (range: 4-8 months), is characterized by an augmentation in the frequency of seizures, often as EPC, and an increase in the degree of hemiparesis. The final residual stage presents with permanent and stable neurological deficits, mostly severe hemiparesis, and a decreased frequency of seizures.
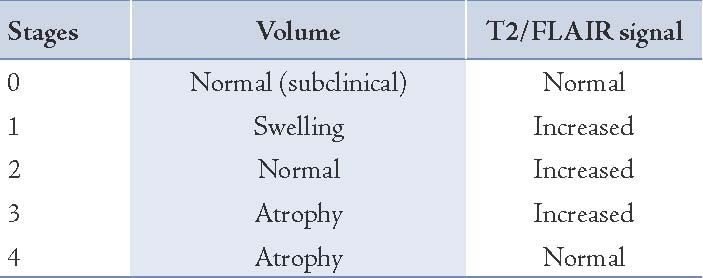
Bien et al. also proposed a five stage MRI model of RE based on a retrospective study of 39 MRI scans of 10 patients (Table 1). The earliest abnormal MRI feature (stage 1) is cortical swelling with hyperintense T2/FLAIR signal with an average duration of 0.3-12.3 months. During the second stage (average duration: 2.1-22.6 months), the features are focal or multifocal T2 and FLAIR hyperintensies involving the cortex or white matter of the unihemisphere, mostly accentuated at the insular and peri-insular region, progressively spreading across the hemisphere. Histopathology, a higher number of T cells and reactive astrocytes corresponding to areas of higher signals can be revealed at this stage. Later on, unihemispheric atrophy (stage 3, average duration: 4.6-103.8 months) sets in, characterized by widening of cortical sulci and dilatation of the ipsilateral lateral ventricle. Our patient presented at this stage. Most of the tissue loss occurs during the first 12 months after the onset of symptomatic disease in the majority of patients. The final phase (stage 4) is characterized by disappearance of the increased signal, leaving a markedly atrophied cerebral hemisphere.
Table 1: MRI stages
Gadolinium-enhanced imaging has not been found to have any added advantage for stablishing the diagnosis even though there are rare reported instances of gadolinium enhancement in RE, associated with an exacerbation of seizure frequency and neurologic deficits.5 The role of magnetic resonance spectroscopy (MRS) is also not well-established. Decreased N-acetyl aspartate (NAA) and increased or normal choline levels, suggestive of neuronal loss, is seen in RE. The presence of lactate with elevated glutamate/glutamine levels is noted following seizure activity.7 Magnetic resonance angiography is useful in excluding large to medium vessel vasculitis as the cause of signal changes, including extremely rare unilateral Moyamoya disease. The role of CT in diagnosis of RE is inferior to that of MRI even though imaging features are similar. The signal abnormalities appear earlier on MRI.5 Fluorodeoxyglucose-positron emission tomography (FDG-PET) may show epileptogenic foci as area of hypometabolic activity and help towards identifying areas not detected by MRI, especially at early stages of disease.7,8 Hyperactivity on FDG-PET may also be seen during the immediate postictal phase. Single photon emission computed tomography (SPECT) may also show parallel findings and results.8
The differential diagnoses for RE include Dyke-Davidoff-Masson syndrome, Sturge-Weber syndrome, hemimegalencephaly, and unihemispheric cerebral vasculitis. Dyke Davidoff Masson syndrome (DDMS) is a set of conditions of different etiologies, leading to unilateral cerebral atrophy with homolateral calvarial hypertrophy and hyperpneumatization of sinuses. The etiology may be classified into congenital and acquired groups. In the congenital variety, cerebral damage usually has a vascular origin. In the acquired type, cerebral insults occur during perinatal period or later and causes include trauma, intracranial hemorrhage, infection and ischemia; and in premature infants, subependymal germinal matrix and intraventricular hemorrhage. As the insult to brain occurs much earlier in intrauterine or perinatal period compared to RE, there is a compensatory overdevelopment of paranasal sinuses and mastoid air cells, ipsilateral calvarial thickening and elevation of the petrous ridge, sphenoid wing and orbital roof.9 Our patient did not present with any of these features.
The Sturge-Weber syndrome is a rare neurocutaneous syndrome characterized by facial capillary and capillary-venous malformations in trigeminal nerve distribution, leptomeningeal venous angiomatosis, seizures, dementia and hemiplegia. Characteristic imaging findings of the cerebral atrophy with gyral or curvilinear calcification (typically described as tram track), enhancing angiomas and ipsilateral enlarged choroid plexus readily differentiate it from RE. Secondary compensatory skull changes to atrophy as in DDMS are also described, but considered as a separate entity.8,9
Hemimegalencephaly is characterized by a large unihemisphere with ipsilateral ventriculomegaly as a result of partial or complete hamartomatous overgrowth of cerebral hemisphere. The affected hemisphere may show focal or diffuse neuronal migration defects, with areas of polymicrogyria, pachygyria and heterotopia. The frontal horn of the ipsilateral ventricle appears straight and pointed anteriorly and superiorly with an indistinct cortical–white matter junction and variable degrees of T2 hyperintensity of the white matter due to heterotopia and gliosis. Unihemispheric cerebral vasculitis is very rare even though one adult case showing progressive atrophy of a single hemisphere with signal changes and parenchymal gadolinium enhancement has been reported.6 The above differential diagnoses were easily ruled out in our patients based on MRI findings (Fig. 3) and DSA.
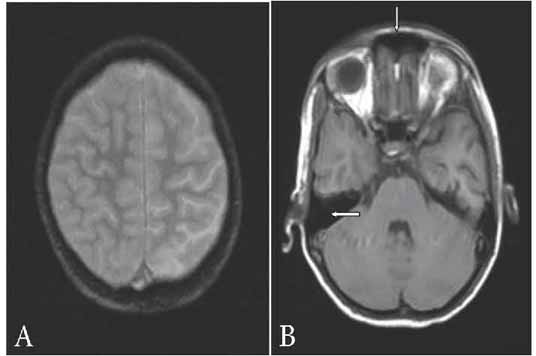
Figure 3: (a) Axial gradient-echo image (TR: 704 ms, TE: 15 ms, slice thickness: 4 mm) showing no abnormal gyral calcification; (b) Axial T1-weighted image (TR: 660 ms, TE: 14 ms, slice thickness: 4 mm) showing normally developed frontal sinus and mastoid air cells.
The outcome of RE is disappointing though early initiation of therapy delays the progression of disease. Treatment includes immunosuppressive and immunomodulator regimens in the form of steroids, immunoglobulins, plasma exchange and tacrolimus. They are useful during the acute stage but may have side effects.10 Our patient was also started on immunomodulatory therapy and did not deteriorate neurologically on follow-up of seven months. However, hemispherectomy has been the most efficient option to eradicate seizures and prevent further deterioration in cognition.11,12
Conclusion
MRI abnormalities of RE evolve from the initial unihemispheric swelling and high signal on T2W and FLAIR images to abnormalities spreading across the affected hemisphere, followed by severe atrophy and disappearance of abnormal signal. In pediatric unihemispheric progressive cerebral atrophy, RE should be considered in the differential diagnosis, especially if there is no intracranial calcification or contrast enhancement. The radiologist plays an active role as imaging is an important tool for early diagnosis and excluding differential diagnoses, which can modify the progression of disease.
Acknowledgements
The authors reported no conflict of interests and no funding was received for this work.
References
1. Rasmussen T, Olszewski J, Lloydsmith D. Focal seizures due to chronic localized encephalitis. Neurology 1958 Jun;8(6):435-445.
2. Bien CG, Bauer J, Deckwerth TL, Wiendl H, Deckert M, Wiestler OD, et al. Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism in Rasmussen’s encephalitis. Ann Neurol 2002a Mar;51(3):311-318.
3. Bien CG, Granata T, Antozzi C, Cross JH, Dulac O, Kurthen M, et al. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis: a European consensus statement. Brain 2005 Mar;128(Pt 3):454-471.
4. Rogers SW, Andrews PI, Gahring LC, Whisenand T, Cauley K, Crain B, et al. Autoantibodies to glutamate receptor GluR3 in Rasmussen’s encephalitis. Science 1994 Jul;265(5172):648-651.
5. Bien CG, Urbach H, Deckert M, Schramm J, Wiestler OD, Lassmann H, et al. Diagnosis and staging of Rasmussen’s encephalitis by serial MRI and histopathology. Neurology 2002b Jan;58(2):250-257.
6. Bien CG, Widman G, Urbach H, Sassen R, Kuczaty S, Wiestler OD, et al. The natural history of Rasmussen’s encephalitis. Brain 2002d Aug;125(Pt 8):1751-1759.
7. Wellard RM, Briellmann RS, Wilson JC, Kalnins RM, Anderson DP, Federico P, et al. Longitudinal study of MRS metabolites in Rasmussen encephalitis. Brain 2004 Jun;127(Pt 6):1302-1312.
8. Rastogi S, Lee C, Salamon N. Neuroimaging in pediatric epilepsy: a multimodality approach. Radiographics 2008 Jul-Aug;28(4):1079-1095.
9. Kochar DK, Jain N, Sharma BV, Kumawat BL, Meena CB. Dyke-Davidoff Masson syndrome : neuroimage. Neurol India 2001 Dec;49(4):417-418.
10. Al-Futaisi A. Childhood multiple sclerosis and related disorders. Oman Med J 2007 Oct;22(3):11-15.
11. Deb P, Sharma MC, Gaikwad S, Tripathi M, Chandra PS, Jain S, et al. Neuropathological spectrum of Rasmussen encephalitis. Neurol India 2005 Jun;53(2):156-160, discussion 160-161.
12. Hart YM, Andermann F, Robitaille Y, Laxer KD, Rasmussen T, Davis R. Double pathology in Rasmussen’s syndrome: a window on the etiology? Neurology 1998 Mar;50(3):731-735.
|