|
Abstract
Angiosarcomas are malignant tumors, which originate from the vessel endothelium and resemble the vessel structure. Stewart-Treves syndrome is an angiosarcoma which in general, develops in female patients after mastectomy and axillary lymph node dissection and is associated with chronic lymphedema. The prognosis of this rare complication is very poor. We present the case of a 52-year-old female who had undergone mastectomy due to breast cancer and 13 years later required shoulder disarticulation due to Stewart-Treves syndrome.
Keywords: Angiosarcoma; Lymphedema; Stewart-Treves syndrome.
Introduction
Angiosarcomas are rare malignant vascular tumors which originate from vascular endothelioma and imitate the vascular structure.1 It is seen most frequently in the breast.2 Angiosarcomas develop many years after the treatment of breast cancer and typically occur in patients with lymphedema who have received radiotherapy. The incidence of angiosarcoma after treatment is 0.16% and sporadic frequency is approximately 1/3200 of this.3 Stewart Treves syndrome is a type of angiosarcoma associated with chronic lymphedema which develops after mastectomy and axillary lymph node dissection. Survey of Stewart Treves syndrome is 10 to 15 months. Five-year survival has been reported as 10% to 33%.1,4
Case Report
A 52-year-old female presented with complaints of red to purplish colored erupted lesions and diffuse swelling and pain in the arm. The patient's history revealed total mastectomy, axillary lymph node dissection and radiotherapy 13 years ago and diffuse edema on the left arm began 4 years after the surgery. The swelling on the arm increased over the past 11 months and red to purple colored eruptions arose on the forearm in the past 2 months. Physical examination determined diffuse edema on the left arm and red to purple erupted lesions located proximally on the left forearm, especially on the extensor surface (Fig. 1). In the radiological examination, bony structures were normal on the X-rays. On MRI, diffuse lymphedema on the forearm and hyperintense multiple nodular masses attached to the muscle in the lymphedema at T1A sections were observed. There were multiple nodular masses with more intermediate intensity than lymphedema at T2A sequences and all were in subcutaneous fatty tissue. Multiple nodules and lesions suggested fascia invasion with the largest being 6.5 × 5.5 cm in size (Fig. 2). Pathological examination revealed vascular and papillary structures with ovoid round hyperchromatic pleomorphic nuclei and narrow pink cytoplasm with a significant nucleolus including erythrocytes in their lumen, which were evaluated as angiosarcoma. (Fig. 3)
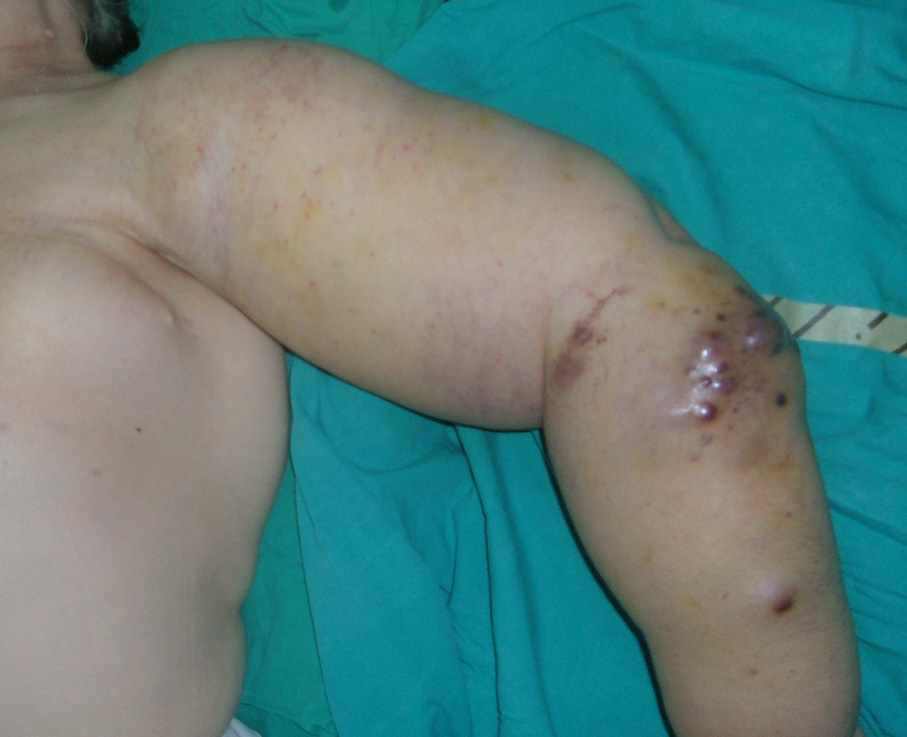
Figure 1: Preoperative clinic image. Red to purple erupted lesions (multiple nodules) located proximally on the left forearm, especially on the extensor surface with diffuse edema.
Upon these results, a radical procedure was considered for the patient and shoulder disarticulation with an anterior approach was performed. On histopathological examination of the amputated limb, angiosarcoma with negative surgical borders was obtained. The patient was discharged fully recovered on postoperative day 5. The patient was on follow-up at the Radiation Oncology and Medical Oncology departments. The patient currently has no complaints at the 25-month follow-up.
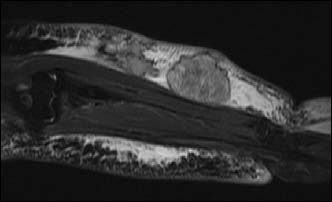
Figure 2: Preoperative MRI findings. Multiple nodules and lesions suggested fascia invasion with the largest being 6.5 × 5.5 cm in size.
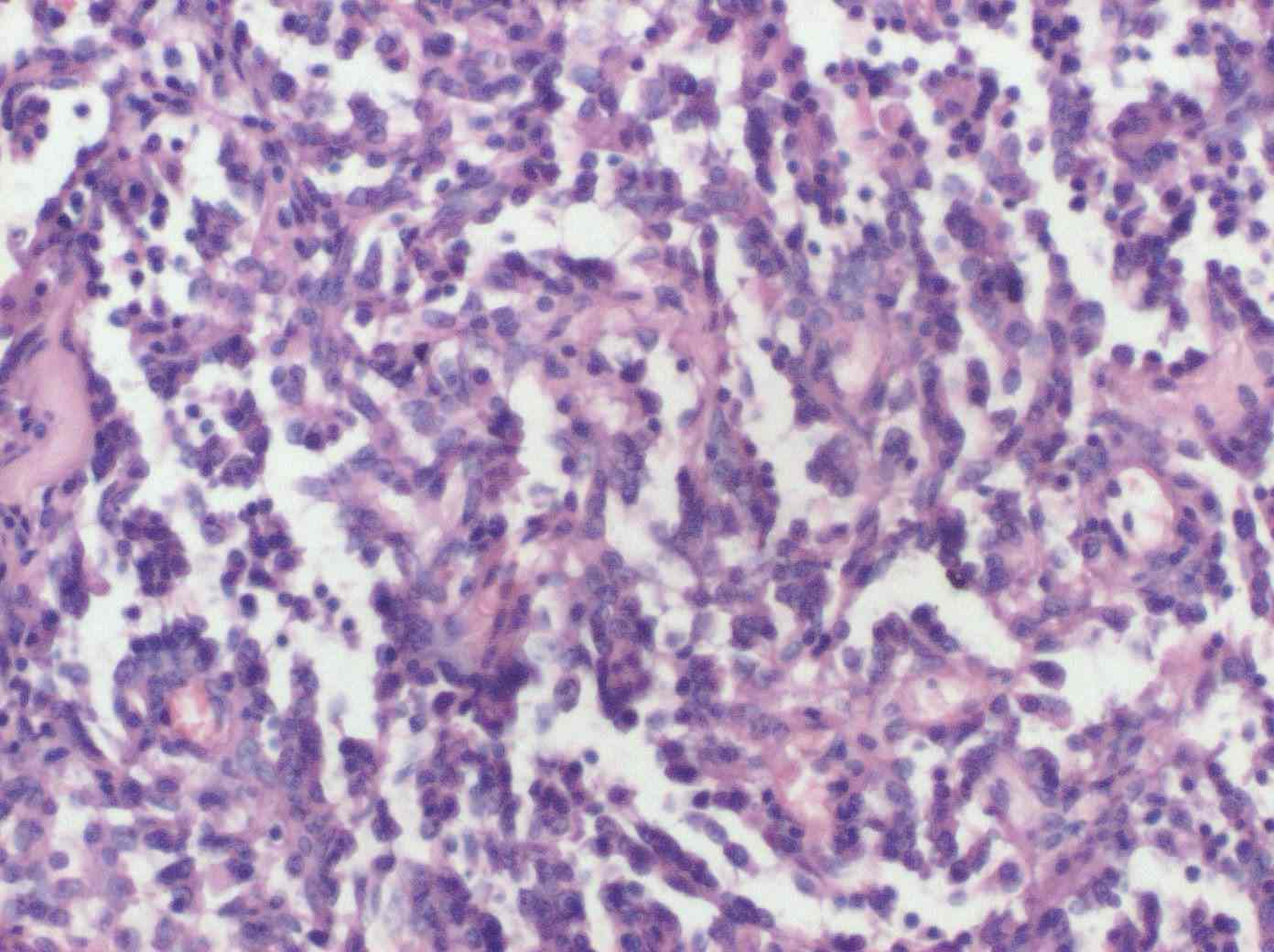
Figure 3: Histologic findings. H&E stain (×200) demonstrated abnormal, pleomorphic, and malignant endothelial cells.
Discussion
Angiosarcomas are malignant tumors in which many functional and morphological features of the epithelial tissue are repeated.5-7 True angiosarcoma is often subcutaneous and aggressive.5,8,9 While well-differentiated forms resemble hemangiomas, it is difficult to differentiate anaplastic types from carcinomas or melanomas. The term angiosarcoma is used for all endothelially differentiated sarcomas, regardless of whether they are vascular or associated with lymphatic endothelium.10
Lymphedema occurs as a result of an increase of protein-rich interstitial fluid when lymphatic drainage is disrupted. Lymphedema deforms local immune response by rarifying movements of immunocompetent cells regardless of the origin. Furthermore, lymphedema causes collateral lymphatic and blood vessel formation by stimulating the angiogenesis in that area. The immunologically weakened area becomes prone to malignant tumors and also vascular tumors due to continuous angiogenic stimulus.11
Angiosarcomas are divided into 3 types. Idiopathic angiosarcoma develops due to chronic lymphedema after mastectomy (STS) and after radiotherapy.1,3 The angiosarcoma which develops after mastectomy was defined by Stewart and Treves in 1948.12 This rare neoplasia develops in less than 1% of mastectomy patients.1,3 Increase in the frequency of angiosarcoma was reported in a parallel increase of the use of radiotherapy for the treatment of breast cancer.13 The age of appearance of angiosarcoma is approximately 60 years and the period after mastectomy to the appearance of the tumor is approximately 10 years.1,4 Angiosarcomas after treatment may be detected after many years on the anterior chest wall of patients who have undergone mastectomy and radiotherapy. Recent studies have reported an increase in the frequency of angiosarcoma in patients on whom lumpectomy had been performed and to whom radiotherapy had been applied.3,14,15 The case presented here was 52 years old and the angiosarcoma developed 13 years after mastectomy. A clinical initiation of angiosarcoma of slow-progressing red to purple blots which will resemble a bruise, injury or hemangioma may occur after any trauma.
There is no standard treatment protocol for angiosarcomas.16 Surgical treatment is generally used in the treatment of primary angiosarcomas, in which the aim should be to obtain clean surgical borders.16,17 Radiotherapy,18 chemotherapy,19,20 angiogenesis inhibitors,21 liposomal chemotherapeutics,21,22 antiangiogenetic treatment,23 and brachytherapy are used as therapy either alone or in combination. Angiosarcoma is a malignancy with a very poor prognosis.24 The amputation results for early diagnosed patients may be promising; however, when the tumor reaches beyond palpable borders clinically, determination of the surgical border may be difficult. The response to chemotherapy is better and it has been reported that survival may be extended if there is a good response to the first treatment.4 Radiotherapy is generally used as a palliative approach.25 The survey after diagnosis of STS is 10 to 15 months.26 Five-year survival after treatment has been reported as 10% to 33%.1,24 In the case presented here, as no distant metastasis was present (M0), shoulder disarticulation was performed. The patient was syndrome free at the 25- month follow-up.
Conclusion
Excision with safe surgical borders in STS has been evaluated as the best treatment option. The size of the tumor and complete excision are the most important factors affecting the prognosis. Therefore, it is important to know and evaluate the clinical findings of this condition (STS) in patients who have undergone mastectomy and to be aware in terms of the diagnosis. Although the case here does not present any new information to literature, the aim was to be a reminder of this rare complication which is difficult to diagnose in the early period. As this complication has a bad prognosis, it is important to leave a safe surgical area to prevent relapses.
Acknowledgements
The authors reported no conflict of interest and no funding was received for this work.
References
1.Calonje E, Mackie RM. Soft-tissue tumours and tumour like conditions, In; Burns T, Breathnach S, Cox N, Griffiths C, editors, Text Book of Dermatology. 7th ed. Oxford; Blackwell Science; 2004, p.53.28-9.
2.Rosen PP. Sarcoma. In: Rosen PP. Rosen’s Breast Pathology. Philadelphia: Lippencott, Williams,&Wilkins; 2001. p, 813-61.
3.Strobbe LJ, Peterse HL, vanTinteren H, et al. Angiosarcoma of the breast after conservation therapy for invasive cancer, the incidence and outcome . an unforeseen sequla. Breast Cancer Res Trea 1198;47:101-9.
4.Noguchi M, Hasegawa H, Tajiri K, De Aretxabala X, Miyazaki I, Terahata S, et al. Stewart-Treves syndrome. A report of two cases with a review of Japanese literature. Jpn J Surg 1987 Sep;17(5):407-412.
5.Khalifa MA, Montgomery EA, Azumi N, Gomes MN, Zeman RK, Min KW, et al. Solitary fibrous tumors: a series of lesions, some in unusual sites. South Med J 1997 Aug;90(8):793-799.
6.Meis-Kindblom JM, Kindblom LG. Angiosarcoma of soft tissue: a study of 80 cases. Am J Surg Pathol 1998 Jun;22(6):683-697.
7.Fletcher CD, Beham A, Bekir S, Clarke AM, Marley NJ. Epithelioid angiosarcoma of deep soft tissue: a distinctive tumor readily mistaken for an epithelial neoplasm. Am J Surg Pathol 1991 Oct;15(10):915-924.
8.Naka N, Ohsawa M, Tomita Y, Kanno H, Uchida A, Aozasa K. Angiosarcoma in Japan. A review of 99 cases. Cancer 1995 Feb;75(4):989-996.
9.Naka N, Ohsawa M, Tomita Y, Kanno H, Uchida A, Myoui A, et al. Prognostic factors in angiosarcoma: a multivariate analysis of 55 cases. J Surg Oncol 1996 Mar;61(3):170-176.
10.Schmitz-Rixen T, Horsch S, Arnold G, Peters PE. Angiosarcoma in primary lymphedema of the lower extremity–Stewart-Treves syndrome. Lymphology 1984 Jun;17(2):50-53.
11. Ruocco V, Schwartz RA, Ruocco E. Lymphedema: an immunologically vulnerable site for development of neoplasms. J Am Acad Dermatol 2002 Jul;47(1):124-127.
12.Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema; a report of six cases in elephantiasis chirurgica. Cancer 1948 May;1(1):64-81.
13.Hanasono MM, Osborne MP, Dielubanza EJ, Peters SB, Gayle LB. Radiation-induced angiosarcoma after mastectomy and TRAM flap breast reconstruction. Ann Plast Surg 2005 Feb;54(2):211-214.
14.Monroe AT, Feigenberg SJ, Mendenhall NP. Angiosarcoma after breast-conserving therapy. Cancer 2003 Apr;97(8):1832-1840.
15.Rubino C, Shamsaldin A, Lê MG, Labbé M, Guinebretière JM, Chavaudra J, et al. Radiation dose and risk of soft tissue and bone sarcoma after breast cancer treatment. Breast Cancer Res Treat 2005 Feb;89(3):277-288.
16.Ocaña A, Delgado C, Rodríguez CA, Bellido L, Izquierdo N, Martín R, et al. Case 3. Upper limb lymphangiosarcoma following breast cancer therapy. J Clin Oncol 2006 Mar;24(9):1477-1478.
17.Kaufmann T, Chu F, Kaufman R. Post-mastectomy lymphangiosarcoma (Stewart-Treves syndrome): report of two long-term survivals. Br J Radiol 1991 Sep;64(765):857-860.
18. Mark RJ, Poen JC, Tran LM, Fu YS, Juillard GF. Angiosarcoma. A report of 67 patients and a review of the literature. Cancer 1996 Jun;77(11):2400-2406.
19. Meis-Kindblom JM, Kindblom LG. Angiosarcoma of soft tissue: a study of 80 cases. Am J Surg Pathol 1998 Jun;22(6):683-697.
20. Budd GT. Management of angiosarcoma. Curr Oncol Rep 2002 Nov;4(6):515-519.
21.Skubitz KM, Haddad PA. Paclitaxel and pegylated-liposomal doxorubicin are both active in angiosarcoma. Cancer 2005 Jul;104(2):361-366.
22. Goetze S, Schmook T, Audring H, Ziegenbein C, Worm M, Schulze P. [Successful treatment of Stewart-Treves syndrome with liposomal doxorubicin]. J Dtsch Dermatol Ges 2004 Jan;2(1):49-52.
23.Vogt T, Hafner C, Bross K, Bataille F, Jauch KW, Berand A, et al. Antiangiogenetic therapy with pioglitazone, rofecoxib, and metronomic trofosfamide in patients with advanced malignant vascular tumors. Cancer 2003 Nov;98(10):2251-2256.
24. Mankin HJ, Hornicek FJ. Diagnosis, classification, and management of soft tissue sarcomas. Cancer Control 2005 Jan-Feb;12(1):5-21.
25.DeLaney TF, Trofimov AV, Engelsman M, Suit HD. Advanced-technology radiation therapy in the management of bone and soft tissue sarcomas. Cancer Control 2005 Jan-Feb;12(1):27-35.
26. Komorowski AL, Wysocki WM, Mituś J. [Stewart-Treves syndrome]. Pol Merkur Lekarski 2004 May;16(95):493-494.
|